
 当前位置:
当前位置:
当前位置:
当前位置:
上传日期:2023-04-21 浏览次数:次
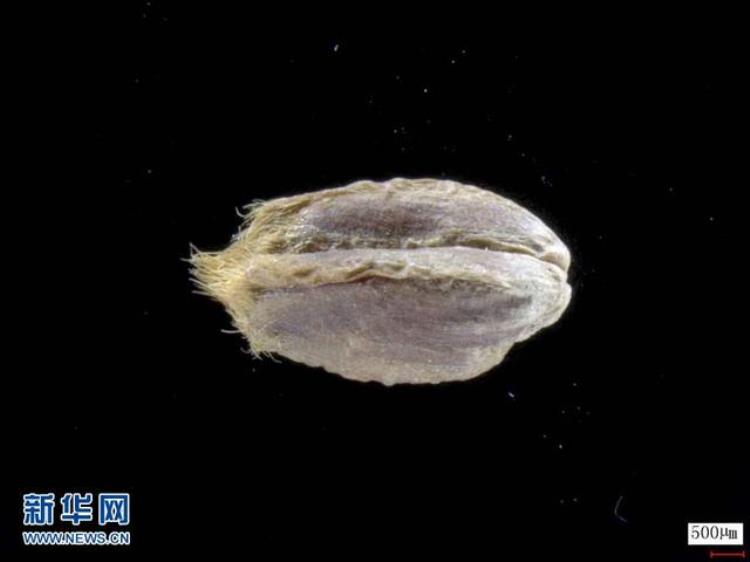 这是距今3800年的小麦种子的超景深三维显微镜照片(吉林大学生命科学学院、吉林大学边疆考古研究中心教授崔银秋提供)。
这是距今3800年的小麦种子的超景深三维显微镜照片(吉林大学生命科学学院、吉林大学边疆考古研究中心教授崔银秋提供)。
小麦进入中国后是如何传播扩散的。中国科学家近期在古代小麦研究领域取得重大进展,成功测定并分析了首例距今约3800年的小麦全基因组序列,证明了古代小麦与中国西南地区现存的普通小麦地方品种的密切关系,提出普通小麦从青藏高原边缘到长江流域的扩散路线。
这项研究系古代小麦基因组研究的首次尝试,为理解东西方文化交流以及农业传播提供了跨时间维度的直接证据。
吉林大学生命科学学院、吉林大学边疆考古研究中心教授崔银秋介绍,六倍体普通小麦是世界上最重要的粮食作物之一。普通小麦驯化始于大约1万年前近东的新月沃地,然后向西扩散到欧洲,向东扩散到东亚。然而,小麦进入中国的传播途径仍不清楚。
为揭开未解之谜,研究人员克服了古植物中DNA含量极低、降解损伤严重的困难,从新疆文物考古研究所提供的距今3800年的单粒小麦种子中成功提取到其基因组DNA,并利用新一代测序技术对中国新疆小河和古墓沟墓地出土的7粒古代小麦种子进行了全基因组测序和序列组装。
崔银秋介绍,基因组数据分析和形态学观察均证明出土的小麦为六倍体普通小麦;通过与来自中国以及世界各地的现代六倍体小麦品种的RNA-seq(转录组测序)数据进行系统发育分析,证明了古代小麦与中国西南地区现存的普通小麦地方品种的密切关系。
研究发现,青藏高原现代地方品种与古代小麦高度相似的等位基因频率,为小麦向高原传播的西南途径提供了更加直接以及有力的分子依据。同时,该项研究还提出了普通小麦从青藏高原边缘到长江流域的扩散路线。
专家认为,该研究结果为中国种植的现存小麦地方品种的栽培起源、扩散和遗传改良提供了重要信息。
上述项目是崔银秋教授研究团队与东北师范大学宫磊教授研究团队合作的成果,得到中国国家自然科学基金等资金资助。
(来源:新华社)

争分夺秒的科研竞争背后,关乎一场算力的角逐 作者 胡珉琦
古DNA研究的力量
古DNA是指从古人类和动植物遗骸以及古生物化石中提取的古生物分子。如果能从古老的化石和考古样本中获取到最原始的古DNA信息,与现代人类和动植物的遗传信息进行比较,就可以为人类、动植物起源与迁徙、文明传播与碰撞、 历史 争议问题给出直接的答案。
20世纪80年代,考古学家和分子生物学家把古DNA研究引入到传统考古学,形成了国际考古研究中的前沿领域——分子考古学。作为 科技 考古的重要手段,古DNA研究在解决人类的起源与迁徙、动植物的家养和驯化过程以及农业的起源和早期发展等重大考古学问题上起到了重要作用。
进入21世纪,随着二代测序技术的普及,古DNA研究迎来了一个黄金发展期。中国的科学家们也在分子考古的浪潮里大放异彩。
早在1998年,吉林大学考古学系就与生命科学学院合作,成立了国内首个考古DNA实验室,开展有关古DNA方面的研究工作。如今,古DNA实验室已初步建立了我国边疆地区的古代DNA基因库,有超过万例的古人类、古动植物样本,数量位居全国第一。
据吉林大学边疆考古研究中心副主任蔡大伟介绍,正是有了这些强有力的资源支持,近年来,吉林大学考古学院运用古DNA优势不断创新和突破,在考古领域驱动了许多重要进展。
例如,对丝路沿线不同时期的动物样本开展全基因组分析,重构了家养动物群体交流的时空框架,展现了丝路大通道在东西方文化交流中的更多 历史 细节;测定和分析了首例距今约3800年古小麦全基因组序列,探究现存小麦地方品种的栽培起源、扩散和遗传改良;报道了中国北方55个古代个体的全基因组数据,探讨了新石器时代农业革命以来中国北方地区的人群互动,为探讨中华文明的起源、形成和发展提供重要证据。
制约古DNA研究的难题
尽管古DNA研究进展飞速,但困难依旧不小。
蔡大伟解释,古DNA研究主要分为两大部分。
第一部分是通过实验手段,从古代生物遗骸中把DNA提取出来,并完成扩增过程。在有机体死亡后,其细胞中的遗传物质就即刻开始降解,给DNA提取和扩增带来了极大的阻力。而且,研究还不可避免的会遇到现代基因的“污染”问题。
第二部分则是测序及数据分析。由于古DNA基因序列片段比现代DNA更短,导致古DNA测序比现代DNA也更复杂。以人类为例,人类基因组是由30亿个碱基对构成的,在得到古人类DNA碎片的序列信息后,考古学家需要借助生物信息学的手段将这些片段进行比对、组装,还原成和现代人DNA 一样完整的、高质量的全基因组。
“这个过程就像完成一幅巨型拼图,没有强大的计算机软件和硬件支撑,是不可能完成的任务。”吉林大学生命科学学院副院长、边疆考古研究中心教授崔银秋直言。
蔡大伟指出,早期科研人员通过通用的计算设备,比如通过CPU去做基因组装,结果发现这一过程非常漫长,“一般完成一次古人类的全基因组样本分析需要耗时至少两周”。
“我们希望把尽可能多的时间放在科学问题的分析和对成果的解释上,而不是消耗在对基础数据的处理和计算上。”崔银秋表示,这就亟需高性能计算和人工智能等新一代技术帮助科学家来加速这一分子考古的过程。
AI计算如何为分子考古提速
那么,算力究竟如何才能帮助加速整个基因拼图重构的过程?
浪潮人工智能和高性能产品线总经理刘军以和吉林大学考古DNA实验室的合作为例介绍道,浪潮采用了一套定制化芯片加速方案,加速古生物基因序列的比对和拼装,再用人工智能的方法和手段,帮助科学家找到感兴趣的突变的基因。这套方案可以帮助考古学家在9.64小时内完成全基因组分析,48分钟完成全外显子组分析,相比基于CPU的方案,基因数据处理速度提升了39倍。
“这就意味着我们的科学家可以用原来四十分之一的时间完成古人类全基因组的比对和拼接工作。”刘军强调。
众所周知,由于DNA可以被复制,特别是当它从父母遗传到子女的时候,只要突变不会致命,那么这些突变也会被复制然后传递给下一代。因此,突变通过时间而不断积累,这就使得科学家能够找到遗传进化的特定链条,并且还可以通过积累的突变而估算时间。
“问题是,这些重要的突变基因在哪里?它们在整个 历史 长河中发生了什么样的演化?”刘军坦言,从这样的追寻过程中,我们才能真正清晰地回溯人类是怎么走过这上百万年的演化 历史 的。
“然而,完成拼接的基因组序列非常长,利用传统方法在上面寻找特定基因的变异过程,是极为困难的一件事,就如同大海捞针。”刘军表示,只有在人工智能的计算方法和手段的加持下,才能帮助科学家在基因的海洋里,找出他们最感兴趣的基因、重要的突变基因。
刘军认为,“从这个角度来说,我们是在为这门古老学科创造一个实用工具,就像科学史上的显微镜、望远镜一样”。通过AI计算,服务科学家细致入微地获取过去得不到客观证据,从而实现洞察 历史 的真相。
刘军还特别提到,在AI计算与考古学研究结合的过程中,也反过来打开了计算研究的视野。“我们要向远处看,向深处看,科学的未来需要依靠怎样的计算技术去迎接挑战。”
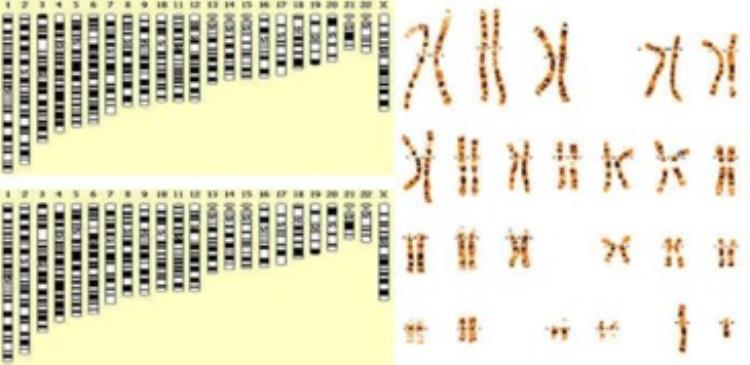
历时22年,完整人类基因组首次被破译,这有哪些重大意义?人类基因组的完全测序是一项重要的科学成就,它为理解人类DNA提供了第一个全面的视角。这些基础信息将增强对人类基因组所有细微功能差异的理解,促进人类疾病的遗传研究。美国国家人类基因组研究中心的科学家们利用新技术研究了世界上第一个完整的、无间隙的人类基因组序列,首次揭示了高度相同的片段,人类基因组中的区域及其变异。这条完整的X染色体核苷酸序列已被完全破译,这将极大地促进人类对生命之书的认识,也将推动人类对各种人类疾病的研究进展。
这有助于回答一些关于染色体如何分离和分裂的基本生物学问题,研究小组还通过使用完整的人类基因组序列发现了超过200万种额外的基因变异。这些研究为622个医学相关基因的基因变异提供了更准确的信息。人类基因组的完整测序是一项重大的科学成就,提供了人类DNA 的第一个完整视图。这些更基本的信息将促进对人类基因组中所有细微功能差异的理解,并将促进人类疾病的基因研究。这是一项规模宏大、跨学科、跨界的科学探索工程,在生命科学领域。目的是确定构成人类染色体的大约60 亿个核苷酸对的序列,即染色体中的DNA 螺旋。
覆盖了基因组中最复杂的一些区域,包括在重要染色体结构中及其周围发现的高度重复的DNA序列,如着丝粒和核糖体DNA。着丝粒是将两条染色体连接在一起的部分,在细胞分裂中起着关键作用。研究团队能够完成一条X染色体的测序,意味着人类具备了克服染色体测序所需的人才、设备和测序方法等多项必要条件。相信很快,研究团队的第n条染色体将继续被测序,直到人类发现隐藏在基因组中的秘密。也许随着科技的发展,在未来的某一天,人类将能够掌握基因组的奥秘,但当人类真正掌握这种奥秘时,人与人之间的差距很可能会迅速拉大,甚至变得不可逾越。
新的间隙填充序列在遗传变异的识别和解释方面也有重大改进,并揭示了以前看不见的着丝粒周围区域的细节。新的研究成果将有助于人们更好地了解人类的进化和生物学特征,并有助于促进衰老、神经退行性疾病、癌症和心脏病等领域的医学研究。

 15930012679
15930012679